Caesium or cesium[note 1] is a chemical element with symbol Cs and atomic number 55. It is a soft, silvery-gold alkali metal with a melting point of 28 °C (82 °F), which makes it one of only five elemental metals that are liquid at or near room temperature.[note 2]Caesium is an alkali metal and has physical and chemical properties similar to those ofrubidium and potassium. The metal is extremely reactive and pyrophoric, reacting with water even at −116 °C (−177 °F). It is the least electronegative element having a stable isotope, caesium-133. Caesium is mined mostly from pollucite, while the radioisotopes, especially caesium-137, a fission product, are extracted from waste produced by nuclear reactors.
Two German chemists, Robert Bunsen and Gustav Kirchhoff, discovered caesium in 1860 by the newly developed method of flame spectroscopy. The first small-scale applications for caesium were as a "getter" in vacuum tubes and in photoelectric cells. In 1967, a specific frequency from the emission spectrum of caesium-133 was chosen to be used in the definition of the second by the International System of Units. Since then, caesium has been widely used in atomic clocks.
Since the 1990s, the largest application of the element has been as caesium formate fordrilling fluids. It has a range of applications in the production of electricity, in electronics, and in chemistry. The radioactive isotope caesium-137 has a half-life of about 30 years and is used in medical applications, industrial gauges, and hydrology. Although the element is only mildly toxic, it is a hazardous material as a metal and its radioisotopes present a high health risk if released into the environment.
Physical properties[edit]
Characteristics
[edit]


High-purity caesium-133 preserved under argon
Caesium is a very soft (it has the lowest hardness of all elements, 0.2 Mohs), veryductile, pale metal, which darkens in the presence of trace amounts of oxygen.[8][9][10]It has a melting point of 28.4 °C (83.1 °F), making it one of the few elemental metals which are liquid near room temperature.Mercury is the only elemental metal with a known melting point lower than caesium.[note 3][12] In addition, the metal has a rather low boiling point, 641 °C (1,186 °F), thelowest of all metals other than mercury.[13] Its compounds burn with a blue[14][15] or violet[15]color.

Secure caesium sample for teaching
Caesium forms alloys with the other alkali metals, as well as with gold, and amalgamswith mercury. At temperatures below 650 °C(1,202 °F), it does not alloy with cobalt, iron,molybdenum, nickel, platinum, tantalum ortungsten. It forms well-defined intermetallic compounds with antimony, gallium, indiumand thorium, which are photosensitive.[8] It mixes with the other alkali metals (except with lithium), and the alloy with a molar distribution of 41% caesium, 47% potassium, and 12%sodium has the lowest melting point of any known metal alloy, at −78 °C (−108 °F).[12][16]A few amalgams have been studied: CsHg
2 is black with a purple metallic luster, while CsHg is golden-colored, also with a metallic luster.[17]
Chemical properties[edit]

Addition of a small amount of caesium to cold water is explosive.
Caesium metal is highly reactive and verypyrophoric. In addition to igniting spontaneously in air, it reacts explosively with water even at low temperatures, more so than other members of the first group of the periodic table.[8] The reaction with solid water occurs at temperatures as low as −116 °C(−177 °F).[12] Because of its high reactivity, the metal is classified as a hazardous material. It is stored and shipped in dry saturated hydrocarbons such as mineral oil. Similarly, it must be handled under inert gassuch as argon. However, a caesium-water explosion is often less powerful than a sodium-water explosion with a similar amount of sodium. This is because caesium explodes instantly upon contact with water, leaving little time for hydrogen to accumulate.[18] Caesium can be stored in vacuum-sealedborosilicate glass ampoules. In quantities of more than about 100 grams (3.5 oz), caesium is shipped in hermetically sealed, stainless steel containers.[8]
The chemistry of caesium is similar to that of other alkali metals, but is more closely similar to that of rubidium, the element above caesium in the periodic table.[19] Some small differences arise from the fact that it has a higher atomic mass and is more electropositivethan other (nonradioactive) alkali metals.[20] Caesium is the most electropositive stable chemical element.[note 4][12] The caesium ion is also larger and less "hard" than those of the lighter alkali metals.
Compounds[edit]

Ball-and-stick model of the cubic coordination of Cs and Cl in CsCl
The vast majority of caesium compounds contain the element as the cation Cs+
, which binds ionically to a wide variety of anions. One noteworthy exception is provided by the caeside anion (Cs−
).[22] Other exceptions include the several suboxides (see section on oxides below).
Returning to more normal compounds, salts of Cs+ are almost invariably colorless unless the anion itself is colored. Many of the simple salts are hygroscopic, but less so than the corresponding salts of the lighter alkali metals. The phosphate,[23] acetate, carbonate, halides, oxide, nitrate, and sulfate salts are water-soluble. Double salts are often less soluble, and the low solubility of caesium aluminium sulfate is exploited in the purification of Cs from its ores. The double salt with antimony (such as CsSbCl
4),bismuth, cadmium, copper, iron, and lead are also poorly soluble.[8]
Caesium hydroxide (CsOH) is hygroscopic and a very strong base.[19] It rapidly etches the surface ofsemiconductors such as silicon.[24] CsOH has been previously regarded by chemists as the "strongest base", reflecting the relatively weak attraction between the large Cs+ ion and OH–.;[14] it is indeed the strongest Arrhenius base, but a number of compounds that cannot exist in aqueous solution, such as n-butyllithium and sodium amide,[19] are more basic.
A stoichiometric mixture of caesium and gold will react to form caesium auride.
Complexes[edit]
Like all metal cations, Cs+ forms complexes with Lewis bases in solution. Because of its large size, Cs+ usually adopts coordination numbers greater than six-coordination, which is typical for the lighter alkali metal cations. This trend is already apparent by the 8-coordination in CsCl, vs the halite motif adopted by the other alkali metal chlorides. Its high coordination number and softness (tendency to form covalent bonds) are the basis of the separation of Cs+ from other cations, as is practiced in the remediation of nuclear wastes, where 137Cs+ is separated from large amounts of nonradioactive K+.[25]
Halides[edit]
Caesium chloride (CsCl) crystallizes in the simple cubic crystal system. Also called the "caesium chloride structure",[20] this structural motif is composed of a primitive cubic lattice with a two-atom basis, each with an eightfold coordination; the chloride atoms lie upon the lattice points at the edges of the cube, while the caesium atoms lie in the holes in the center of the cubes. This structure is shared withCsBr and CsI, and many other compounds that do not contain Cs. In contrast, most other alkaline halides adopt the sodium chloride(NaCl) structure.[20] The CsCl structure is preferred because Cs+ has an ionic radius of 174 pm and Cl−
181 pm.[26]
Oxides[edit]

Cs
11O
3 cluster
More so than the other alkali metals, caesium forms numerous binary compounds with oxygen. When caesium burns in air, the superoxide CsO
2 is the main product.[27] The "normal" caesium oxide (Cs
2O) forms yellow-orange hexagonal crystals,[28] and is the only oxide of the anti-CdCl
2 type.[29] It vaporizes at 250 °C (482 °F), and decomposes to caesium metal and the peroxide Cs
2O
2 at temperatures above400 °C (752 °F).[30] Aside from the superoxide and the ozonide CsO
3,[31][32] several brightly coloredsuboxides have also been studied.[33] These include Cs
7O, Cs
4O, Cs
11O
3, Cs
3O (dark-green[34]), CsO,Cs
3O
2,[35] as well as Cs
7O
2.[36][37] The latter may be heated under vacuum to generate Cs
2O.[29] Binary compounds with sulfur, selenium, and tellurium also exist.[8]
Isotopes[edit]
Main article: Isotopes of caesium
Caesium has a total of 39 known isotopes that range in their mass number (i.e. number of nucleons in its nucleus) from 112 to 151. Several of these are synthesized from lighter elements by the slow neutron capture process (S-process) inside old stars,[38] as well as inside supernova explosions (R-process).[39] However, the only stable isotope is 133Cs, which has 78 neutrons. Although it has a largenuclear spin (7/2+), nuclear magnetic resonance studies can be done with this isotope at a resonating frequency of 11.7 MHz.[40]

Decay of caesium-137
The radioactive 135Cs has a very long half-life of about 2.3 million years, while 137Csand 134Cs have half-lives of 30 and two years, respectively. 137Cs decomposes to a short-lived 137mBa by beta decay, and then to nonradioactive barium, while 134Cs transforms into 134Ba directly. The isotopes with mass numbers of 129, 131, 132 and 136, have half-times between a day and two weeks, while most of the other isotopes have half-lives from a few seconds to fractions of a second. There are at least 21 metastable nuclear isomers. Other than 134mCs (with a half-life of just under 3 hours), all are very unstable and decay with half-lives of a few minutes or less.[41][42]
The isotope 135Cs is one of the long-lived fission products of uranium which form innuclear reactors.[43] However, its fission product yield is reduced in most reactors because its predecessor, 135Xe, is an extremely potent neutron poison and transmutes frequently to stable 136Xe before it can decay to135Cs.[44][45]
Because of its beta decay (to 137mBa), 137Cs is a strong emitter of gamma radiation.[46] Its half-life makes it the principal medium-lived fission product along with 90Sr—both are responsible for radioactivity of spent nuclear fuel after several years of cooling up to several hundred years after use.[47] For example 137Cs together with 90Sr currently generate the largest source of radioactivity generated in the area around the Chernobyl disaster.[48] It is not feasible to dispose of 137Cs through neutron capture (due to the low capture rate) and as a result it must be allowed to decay.[49]
Almost all caesium produced from nuclear fission comes from beta decay of originally more neutron-rich fission products, passing through various isotopes of iodine and of xenon.[50] Because iodine and xenon are volatile and can diffuse through nuclear fuel or air, radioactive caesium is often created far from the original site of fission.[51] With the commencement of nuclear weapons testing around 1945, 137Cs was released into the atmosphere and then returned to the surface of the earth as a component of radioactive fallout.[8]
Occurrence[edit]

Pollucite, a caesium mineral
See also: Caesium minerals
Caesium is a relatively rare element as it is estimated to average approximately 3 parts per million in the Earth's crust.[52] This makes it the 45th most abundant of all elements and the 36th of all the metals. Nevertheless, it is more abundant than such elements as antimony, cadmium, tin and tungsten, and two orders of magnitude more abundant than mercury orsilver, but 3.3% as abundant as rubidium—with which it is so closely chemically associated.[8]
Due to its large ionic radius, caesium is one of the "incompatible elements".[53] Duringmagma crystallization, caesium is concentrated in the liquid phase and crystallizes last. Therefore, the largest deposits of caesium are zone pegmatite ore bodies formed by this enrichment process. Because caesium does not substitute for potassium as readily as does rubidium, the alkali evaporite mineralssylvite (KCl) and carnallite (KMgCl
3·6H
2O) may contain only 0.002% caesium. Consequently, Cs is found in few minerals. Percentage amounts of caesium may be found in beryl (Be
3Al
2(SiO
3)
6) and avogadrite ((K,Cs)BF
4), up to 15 wt% Cs2O in the closely related mineral pezzottaite (Cs(Be2Li)Al2Si6O18), up to 8.4 wt% Cs2O in the rare mineral londonite ((Cs,K)Al
4Be
4(B,Be)
12O
28), and less in the more widespread rhodizite.[8] The only economically important source mineral for caesium is pollucite Cs(AlSi
2O
6), which is found in a few places around the world in zoned pegmatites, and is associated with the more commercially important lithium minerals lepidoliteand petalite. Within the pegmatites, the large grain size and the strong separation of the minerals create high-grade ore for mining.[54]
One of the world's most significant and richest sources of the metal is the Tanco mine at Bernic Lake in Manitoba, Canada. The deposits there are estimated to contain 350,000 metric tons of pollucite ore, which represent more than two-thirds of the world's reserve base.[54][55] Although the stoichiometric content of caesium in pollucite is 42.6%, pure pollucite samples from this deposit contain only about 34% caesium, while the average content is 24 wt%.[55] Commercial pollucite contains over 19% caesium.[56] The Bikita pegmatite deposit in Zimbabwe is mined for its petalite, but it also contains a significant amount of pollucite. Notable amounts of pollucite are also mined in the Karibib Desert, Namibia.[55] At the present rate of world mine production of 5 to 10 metric tons per year, reserves will last for thousands of years.[8]
Production[edit]
The mining of pollucite ore is a selective process and is conducted on a small scale in comparison with most metal mining operations. The ore is crushed, hand-sorted, but not usually concentrated, and then ground. Caesium is then extracted from pollucite mainly by three methods: acid digestion, alkaline decomposition, and direct reduction.[8][57]
In the acid digestion, the silicate pollucite rock is dissolved with strong acids such as hydrochloric (HCl), sulfuric (H
2SO
4), hydrobromic(HBr), or hydrofluoric (HF) acids. With hydrochloric acid, a mixture of soluble chlorides is produced, and the insoluble chloride double salts of caesium are precipitated as caesium antimony chloride (Cs
4SbCl
7), caesium iodine chloride (Cs
2ICl), or caesium hexachlorocerate (Cs
2(CeCl
6)). After separation, the pure precipitated double salt is decomposed, and pure CsCl is obtained after evaporating the water. The method using sulfuric acid yields the insoluble double salt directly as caesium alum (CsAl(SO
4)
2·12H
2O). The aluminium sulfate in it is converted to the insoluble aluminium oxide by roasting the alum with carbon, and the resulting product isleached with water to yield a Cs
2SO
4 solution.[8]
The roasting of pollucite with calcium carbonate and calcium chloride yields insoluble calcium silicates and soluble caesium chloride. Leaching with water or dilute ammonia (NH
4OH) yields then a dilute chloride (CsCl) solution. This solution can be evaporated to produce caesium chloride or transformed into caesium alum or caesium carbonate. Albeit not commercially feasible, direct reduction of the ore with potassium, sodium or calcium in vacuum can produce caesium metal directly.[8]
Most of the mined caesium (as salts) is directly converted into caesium formate (HCOO−Cs+) for applications such as oil drilling. To supply the developing market, Cabot Corporation built a production plant in 1997 at the Tanco mine near Bernic Lake in Manitoba, with a capacity of 12,000 barrels (1,900 m3) per year of caesium formate solution.[58] The primary smaller-scale commercial compounds of caesium are caesium chloride and its nitrate.[59]
Alternatively, caesium metal may be obtained from the purified compounds derived from the ore. Caesium chloride, and the other caesium halides, as well, can be reduced at 700 to 800 °C (1,292 to 1,472 °F) with calcium or barium, followed by distillation of the caesium metal. In the same way, the aluminate, carbonate, or hydroxide may be reduced by magnesium.[8] The metal can also be isolated by electrolysis of fused caesium cyanide (CsCN). Exceptionally pure and gas-free caesium can be made by the thermal decomposition at 390 °C (734 °F) of caesium azide CsN
3, which is produced from aqueous caesium sulfate and barium azide.[57] In vacuum applications, caesium dichromate can be reacted with zirconium forming pure caesium metal without other gaseous products.[59]Cs
2Cr
2O
7 + 2 Zr → 2 Cs + 2 ZrO
2+ Cr
2O
3
The price of 99.8% pure caesium (metal basis) in 2009 was about US$10 per gram ($280 per ounce), but its compounds are significantly cheaper.[55]
History[edit]

Gustav Kirchhoff (left) and Robert Bunsen (center) discovered caesium spectroscopically.
In 1860, Robert Bunsen and Gustav Kirchhoff discovered caesium in the mineral water fromDürkheim, Germany. Due to the bright blue lines in its emission spectrum, they chose a name derived from the Latin word caesius, meaning sky-blue.[note 5][60][61][62] Caesium was the first element to be discovered spectroscopically, only one year after the invention of thespectroscope by Bunsen and Kirchhoff.[12]
To obtain a pure sample of caesium, 44,000 litres (9,700 imp gal; 12,000 US gal) of mineral water had to be evaporated to yield 240 kilograms (530 lb) of concentrated salt solution. Thealkaline earth metals were precipitated either as sulfates or oxalates, leaving the alkali metal in the solution. After conversion to the nitrates and extraction with ethanol, a sodium-free mixture was obtained. From this mixture, the lithium was precipitated by ammonium carbonate. Potassium, rubidium and caesium form insoluble salts with chloroplatinic acid, but these salts show a slight difference in solubility in hot water. Therefore, the less-soluble caesium and rubidium hexachloroplatinate ((Cs,Rb)2PtCl6) could be obtained by fractional crystallization. After reduction of the hexachloroplatinate with hydrogen, caesium and rubidium could be separated by the difference in solubility of their carbonates in alcohol. The process yielded 9.2 grams (0.32 oz) of rubidium chloride and 7.3 grams (0.26 oz) of caesium chloride from the initial 44,000 liters of mineral water.[61]
The two scientists used the caesium chloride thus obtained to estimate the atomic weight of the new element at 123.35 (compared to the currently accepted one of 132.9).[61] They tried to generate elemental caesium by electrolysis of molten caesium chloride, but instead of a metal, they obtained a blue homogenous substance which "neither under the naked eye nor under the microscope" showed the slightest trace of metallic substance"; as a result, they assigned it as a subchloride (Cs
2Cl). In reality, the product was probably a colloidalmixture of the metal and caesium chloride.[63] The electrolysis of the aqueous solution of chloride with a mercury anode produced a caesium amalgam which readily decomposed under the aqueous conditions.[61] The pure metal was eventually isolated by the German chemist Carl Setterberg while working on his doctorate with Kekulé and Bunsen.[62] In 1882, he produced caesium metal by electrolyzing caesium cyanide, and thus avoiding the problems with the chloride.[64]
Historically, the most important use for caesium has been in research and development, primarily in chemical and electrical fields. Very few applications existed for caesium until the 1920s, when it came to be used in radio vacuum tubes. It had two functions; as a getter, it removed excess oxygen after manufacture, and as a coating on the heated cathode, it increased its electrical conductivity. Caesium did not become recognized as a high-performance industrial metal until the 1950s.[65] Applications of nonradioactive caesium includedphotoelectric cells, photomultiplier tubes, optical components of infrared spectrophotometers, catalysts for several organic reactions, crystals for scintillation counters, and in magnetohydrodynamic power generators.[8]
Since 1967, the International System of Measurements has based its unit of time, the second, on the properties of caesium. The International System of Units (SI) defines the second as 9,192,631,770 cycles of the radiation, which corresponds to the transition between two hyperfine energy levels of the ground state of the caesium-133 atom.[66] The 13th General Conference on Weights and Measures of 1967 defined a second as: "the duration of 9,192,631,770 cycles of microwave light absorbed or emitted by the hyperfine transition of caesium-133 atoms in their ground state undisturbed by external fields".
Applications[edit]
Petroleum exploration[edit]
The largest current end-use of nonradioactive caesium is in caesium formate-based drilling fluids for the extractive oil industry.[8]Aqueous solutions of caesium formate (HCOO–Cs+)—made by reacting caesium hydroxide with formic acid—were developed in the mid-1990s for use as oil well drilling and completion fluids. The function of caesium formate as a drilling fluid is to lubricate drill bits, to bring rock cuttings to the surface, and to maintain pressure on the formation during drilling of the well. As completion fluid, which assists the emplacement of control hardware after drilling but prior to production, the function of caesium formate is to maintain the pressure.[8]
The high density of the caesium formate brine (up to 2.3 g·cm−3, or 19.2 pounds per gallon),[67] coupled with the relatively benign nature of most caesium compounds, reduces the requirement for toxic high-density suspended solids in the drilling fluid—a significant technological, engineering and environmental advantage. Unlike the components of many other heavy liquids, caesium formate is relatively environment-friendly.[67] The caesium formate brine can be blended with potassium and sodium formates to decrease the density of the fluids down to that of water (1.0 g·cm−3, or 8.3 pounds per gallon). Furthermore, it is biodegradable and reclaimable, and may be recycled, which is important in view of its high cost (about $4,000 per barrel in 2001).[68] Alkali formates are safe to handle and do not damage the producing formation or downhole metals as their corrosive alternative, high-density brines (such as zinc bromideZnBr
2 solutions),sometimes do; they also require less cleanup and disposal costs.[8]
Atomic clocks[edit]

Atomic clock ensemble at the U.S. Naval Observatory

FOCS-1, a continuous cold caesium fountain atomic clock in Switzerland, started operating in 2004 at an uncertainty of one second in 30 million years
Caesium-based atomic clocks observe electromagnetic transitions in the hyperfine structure of caesium-133 atoms and use it as a reference point. The first accurate caesium clock was built by Louis Essen in 1955 at the National Physical Laboratory in the UK.[69] Since then, they have been improved repeatedly over the past half-century, and form the basis for standards-compliant time and frequency measurements. These clocks measure frequency with an error of 2 to 3 parts in 1014, which would correspond to a time measurement accuracy of 2 nanoseconds per day, or one second in 1.4 million years. The latest versions are accurate to better than 1 part in 1015, which means they would be off by about 2 seconds since the extinction of the dinosaurs65 million years ago,[8] and has been regarded as "the most accurate realization of a unit that mankind has yet achieved."[66]
Caesium clocks are also used in networks that oversee the timing of cell phone transmissions and the information flow on the Internet.[70]
Electric power and electronics[edit]
Caesium vapor thermionic generators are low-power devices that convert heat energy to electrical energy. In the two-electrode vacuum tube converter, it neutralizes the space charge that builds up near the cathode, and in doing so, it enhances the current flow.[71]
Caesium is also important for its photoemissive properties by which light energy is converted to electron flow. It is used in photoelectric cells because caesium-based cathodes such as the intermetallic compound K
2CsSb have low threshold voltage for emission of electrons.[72]The range of photoemissive devices using caesium include optical character recognitiondevices, photomultiplier tubes, and video camera tubes.[73][74] Nevertheless, germanium, rubidium, selenium, silicon, tellurium, and several other elements can substitute caesium in photosensitive materials.[8]
Caesium iodide (CsI), bromide (CsBr) and caesium fluoride (CsF) crystals are employed for scintillators in scintillation counters widely used in mineral exploration and particle physics research, as they are well-suited for the detection of gamma and X-ray radiation. Caesium, being a heavy element, provides good stopping power, contributing to better detectivity. Caesium compounds may also provide a faster response (CsF) and be less hygroscopic (CsI).
Caesium vapor is used in many common magnetometers.[75] The element is also used as an internal standard in spectrophotometry.[76]Like other alkali metals, caesium has a great affinity for oxygen and is used as a "getter" in vacuum tubes.[77] Other uses of the metal include high-energy lasers, vapor glow lamps, and vapor rectifiers.[8]
Centrifugation fluids[edit]
Because of their high density, solutions of caesium chloride, caesium sulfate, and caesium trifluoroacetate (Cs(O
2CCF
3)) are commonly used in molecular biology for density gradient ultracentrifugation.[78] This technology is primarily applied to the isolation of viral particles, subcellular organelles and fractions, and nucleic acids from biological samples.[79]
Chemical and medical use[edit]

A sample of caesium chloride
Relatively few chemical applications exist for caesium.[80] Doping with caesium compounds is used to enhance the effectiveness of several metal-ion catalysts used in the production of chemicals, such as acrylic acid, anthraquinone, ethylene oxide, methanol, phthalic anhydride, styrene, methyl methacrylate monomers, and various olefins. It is also used in the catalytic conversion of sulfur dioxide into sulfur trioxide in the production of sulfuric acid.[8]
Caesium fluoride enjoys niche use in organic chemistry as a base,[19] or as an anhydroussource of fluoride ion.[81] Caesium salts sometimes replace potassium or sodium salts inorganic synthesis, such as cyclization, esterification, and polymerization.
Nuclear and isotope applications[edit]
Caesium-137 is a very common radioisotope used as a gamma-emitter in industrial applications. Its advantages include a half-life of roughly 30 years, its availability from thenuclear fuel cycle, and having 137Ba as stable end product. The high water solubility is a disadvantage which makes it incompatible with large pool irradiators for food and medical supplies.[82] It has been used in agriculture, cancer treatment, and the sterilization of food, sewage sludge, and surgical equipment.[8][83] Radioactive isotopes of caesium in radiation devices were used in the medical field to treat certain types of cancer,[84]but emergence of better alternatives and the use of water-soluble caesium chloride in the sources, which could create wide-ranging contamination, gradually put some of these caesium sources out of use.[85][86] Caesium-137 has been employed in a variety of industrial measurement gauges, including moisture, density, leveling, and thickness gauges.[87] It has also been used in well loggingdevices for measuring the electron density of the rock formations, which is analogous to the bulk density of the formations.[88]
Isotope 137 has also been used in hydrologic studies analogous to those using tritium. It is a daughter product of nuclear fission reactions. With the commencement of nuclear testing around 1945, and continuing through the mid-1980s, caesium-137 was released into the atmosphere, where it is absorbed readily into solution. Known year-to-year variation within that period allows correlation with soil and sediment layers. Caesium-134, and to a lesser extent caesium-135, have also been used in hydrology as a measure of caesium output by the nuclear power industry. While they are less prevalent than either caesium-133 or caesium-137, these isotopes have the advantage of being produced solely from anthropogenic sources.[89]
Other uses[edit]

Schematics of an electrostatic ion thruster which was initially developed for use with caesium or mercury
Caesium and mercury were used as a propellant in early ion engines designed for spacecraft propulsion on very long interplanetary or extraplanetary missions. The ionization method was to strip the outer electron from the propellant upon contact with a tungsten electrode that had voltage applied. Concerns about the corrosive action of caesium on spacecraft components have pushed development in the direction of use of inert gas propellants such as xenon; this is easier to handle in ground-based tests and has less potential to interfere with the spacecraft.[8] Eventually, xenon was used in the experimental spacecraft Deep Space 1 launched in 1998.[90][91] Nevertheless, field emission electric propulsion thrusters which use a simple system of accelerating liquid metal ions such as of caesium to create thrust have been built.[92]
Caesium nitrate is used as an oxidizer and pyrotechnic colorant to burn siliconin infrared flares[93] such as the LUU-19 flare,[94] because it emits much of its light in the near infrared spectrum.[95] Caesium has been used to reduce theradar signature of exhaust plumes in the SR-71 Blackbird military aircraft.[96]Caesium, along with rubidium, has been added as a carbonate to glass because it reduces electrical conductivity and improves stability and durability of fiber optics and night vision devices. Caesium fluoride or caesium aluminium fluoride are used in fluxes formulated for the brazing of aluminium alloys that contain magnesium.[8]
Prognostications[edit]
Magnetohydrodynamic (MHD) power-generating systems were researched, but failed to gain widespread acceptance.[97] Caesium metal has also been considered as the working fluid in high-temperature Rankine cycle turboelectric generators.[98] Caesium salts have been evaluated as antishock reagents to be used following the administration of arsenical drugs. Because of their effect on heart rhythms, however, they are less likely to be used than potassium or rubidium salts. They have also been used to treat epilepsy.[8]
Health and safety hazards[edit]
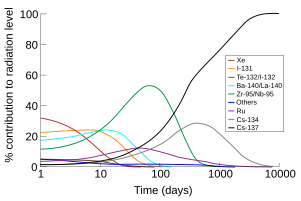
The portion of the total radiation dose (in air) contributed by each isotope versus time after the Chernobyl disasterdepicting caesium-137 becoming the largest source of radiation about 200 days after the accident.[99]
Caesium compounds are rarely encountered by most people, but most are mildly toxic because of chemical similarity of caesium to potassium. Exposure to large amounts of caesium compounds can cause hyperirritability andspasms, but as such amounts would not ordinarily be encountered in natural sources, caesium is not a major chemical environmental pollutant.[100] Themedian lethal dose (LD50) value for caesium chloride in mice is 2.3 g per kilogram, which is comparable to the LD50 values of potassium chloride andsodium chloride.[101]
NFPA 704

3
4
3
W
The fire diamondhazard sign for caesium metal
Caesium metal is one of the most reactive elements and is highly explosive when it comes in contact with water. The hydrogen gas produced by the reaction is heated by the thermal energy released at the same time, causing ignition and a violent explosion. This can occur with other alkali metals, but caesium is so potent that this explosive reaction can even be triggered by cold water.[8] Theautoignition temperature of caesium is also −116 °C, so it is highly pyrophoric, and ignites explosively in air to form caesium hydroxide and various oxides. Caesium hydroxide is a very strong base, and will rapidly corrode glass.[13]
The isotopes 134 and 137 are present in the biosphere in small amounts from human activities and represent a radioactivity burden which varies depending on location. Radiocaesium does not accumulate in the body as effectively as many other fission products (such as radioiodine and radiostrontium). About 10% of absorbed radiocaesium washes out of the body relatively quickly in sweat and urine. The remaining 90% has a half-life between 50 and 150 days.[102] Radiocaesium follows potassium and tends to accumulate in plant tissues, including fruits and vegetables.[103][104][105] It is also well-documented that mushrooms from contaminated forests accumulate radiocaesium (caesium-137) in their fungal sporocarps.[106] Accumulation of caesium-137 in lakes has been a high concern after the Chernobyl disaster.[107][108] Experiments with dogs showed that a single dose of 3.8 millicuries (140 MBq, 4.1 μg of caesium-137) per kilogram is lethal within three weeks;[109] smaller amounts may cause infertility and cancer.[110] TheInternational Atomic Energy Agency and other sources have warned that radioactive materials, such as caesium-137, could be used in radiological dispersion devices, or "dirty bombs"
























